Sampling fungal communities
Sampling fungal communities by environmental DNA
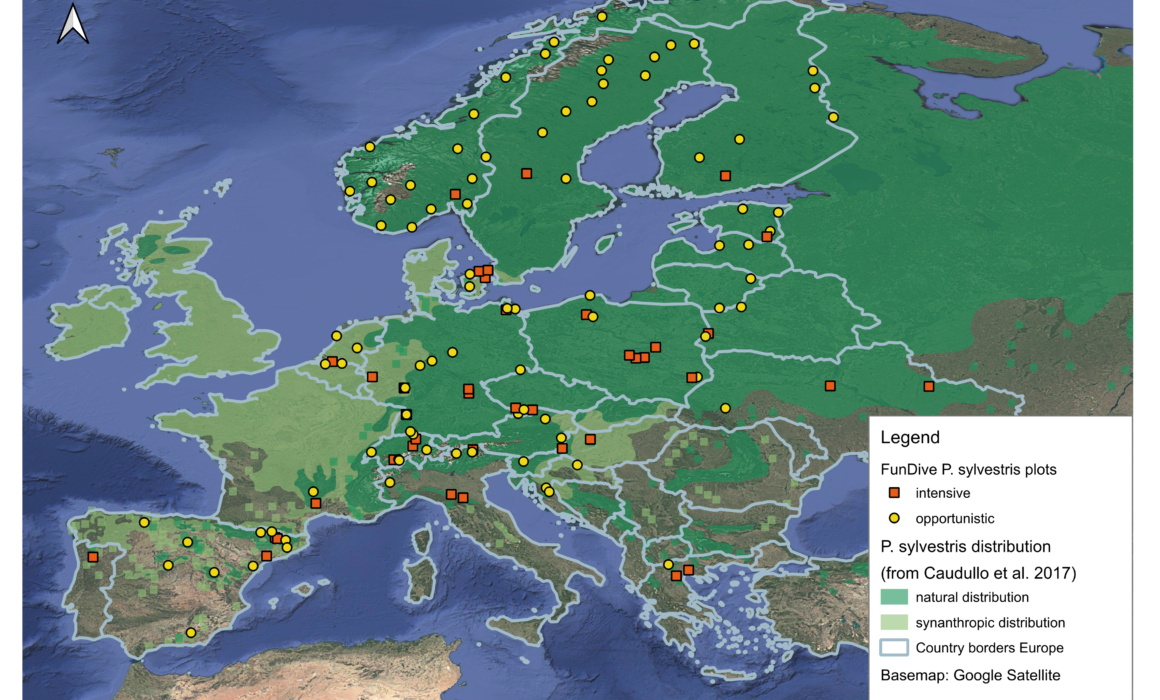
High throughput sequencing of eDNA (DNA metabarcoding) has become a standard technique for characterising fungal communities. The technique holds great potential to resolve fungal conservation challenges, as it neither requires detectable fruit bodies nor taxonomic field expertise. Yet, in order to fully exploit its potential, the method must be optimised specifically for questions relevant for fungal conservation, which so far has mainly been based on sporocarp data. In WP4, we will establish ~120 study plots in forests dominated by Scots pine across Europe. We will collect eDNA samples from soil and deadwood from all the plots, and in 40 plots we will additionally collect DNA from air and conduct traditional fruit body surveys in parallel to analyse strengths and drawbacks of the different approaches. The unique dataset will further yield insights into diversity, uniqueness and conservation value of the different forests and help practitioners to guide fungal conservation. We will also analyze soil and deadwood samples from plots dominated by two other pine species: the black pine (P. nigra) and the stone pine (P. cembra) and compare fungal diversity from these species with the diversity associated with the Scots pine.
Objectives
How can we best capture fungal diversity? We will explore how much of the fungal diversity can be detected by eDNA barcoding versus sporocarp/thallus sampling at plot level, and the overlap among the two approaches, with special reference to conservation relevant species.
Which substrate types should we sample by eDNA to best capture fungal diversity? We will compare the ability of eDNA sampling of various substrate types as tools to capture fungal biodiversity in forest habitats, with a focus on conservation relevant species. We hypothesise that each substrate will give unique insights to fungal diversity, but with considerable variation in the relative contribution to total revealed diversity, depending on forest type.
Which metabarcoding method should we use to best capture fungal diversity? We will combine short- and long-read sequencing pipelines to compare their cost-effectiveness and ability to reliably capture fungal diversity. We hypothesise that overall fungal diversity is best captured with short-read, whereas taxonomic assignments and phylogenetic placements are more precise using long read methods .
What are the patterns of fungal alpha diversity, uniquity, and phylogenetic diversity across European forests? Based on the full dataset we will explore fungal biodiversity patterns across the forest types sampled. We will focus especially on threatened fungi and uniquity patterns to identify plots and forest types with unique value for fungal conservation in Europe. This work will be tightly coordinated with WP5.
Tasks
Establish study plots. We will establish around 120 study plots (50 m2) in Scots pine forests, covering an extensive environmental gradient ranging from subarctic pine forests in Northern Norway, over temperate, boreal and alpine forest in Western and Central Europe to Mediterranean forests spanning from Portugal to Greece.
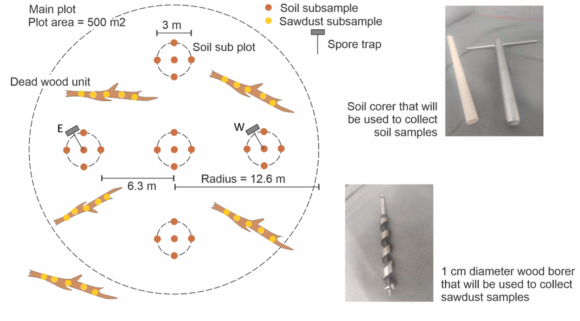 Obtain environmental samples. We will in each plot collect different substrates for metabarcoding analyses, including: (i) soil, (ii) deadwood in all plots, and in 40 plots additionally (iii) wood dwelling fruit bodies, (iv) soil dwelling fruit bodies and (v) aerial spore samples. The latter two sample types (iv and v) will be collected every two weeks, spanning a ten-week-period. The former three sample types (i-iii) will represent pools of numerous sub-samples systematically obtained per plots, in order to cover the microscale fungal diversity.
Obtain environmental samples. We will in each plot collect different substrates for metabarcoding analyses, including: (i) soil, (ii) deadwood in all plots, and in 40 plots additionally (iii) wood dwelling fruit bodies, (iv) soil dwelling fruit bodies and (v) aerial spore samples. The latter two sample types (iv and v) will be collected every two weeks, spanning a ten-week-period. The former three sample types (i-iii) will represent pools of numerous sub-samples systematically obtained per plots, in order to cover the microscale fungal diversity.
DNA library preparation and sequencing. All samples will be frozen upon sampling and later sent to centralized labs for DNA extraction. Library preparation of the fungal ITS metabarcoding marker will be conducted in one lab for all environmental substrates (soil, deadwood, air). The ITS libraries will be analysed using short-read Illumina sequencing, and state-of-the art bioinformatics approaches will be used to produce fungal OTU-matrices that will be taxonomically annotated. DNA extracts from all samples will also be sent to another lab (UiO), where long-read sequencing of the entire rDNA operon will be conducted using circular PacBio-sequencing. Corresponding OTU-matrices will be produced from the long-read sequences, as well as comprehensive sequence alignments for phylogenetic analyses.
Analyse substrate differences. Assess which substrate types best capture fungal diversity / threatened species in different European forests, and what are the overlap in fungal diversity among substrate types.
Compare short and long-read sequences. Assess to what extent long-read sequencing can reveal the same fungal diversity as short read sequencing, and to what degree long-read sequencing provides improved taxonomic annotation.
Analyse eDNA vs sporocarp diversity. We will compare how much of the fungal diversity can be detected through sporocarp/thallus surveys versus eDNA, and evaluate to what degree threatened fungi can be targeted with the two sequencing approaches, versus sporocarps/thalli.
Compare fungal diversity patterns across European forests. In order to better understand the distribution of European threatened fungi.

Taking soil samples in a beautiful Estonian pine forest 
The plot dimensions are measured precisely before sampling (Denmark) 
A close-up of the spore collectors (Switzerland) 
Taking the deadwood sample (Estonia) 
Measuring the length of the deadwood (Poland) 
Beautiful study plot in Czech Republic